Fabry Disease
Fabry Disease is a lysosomal storage disorder inherited in an X-linked recessive manner, caused by a mutation in the GLA gene. As a result of this mutation, there is a deficiency of the enzyme alpha-galactosidase A, which is responsible for breaking down fatty substances. This leads to the accumulation of globotriaosylceramide in various organs.
The Fabry Disease belongs to the group of ultra-rare mucopolysaccharidoses. The condition can cause severe episodes of excruciating, burning pain in the distal parts of the limbs, radiating to different parts of the body.
Characteristic symptoms also include thermoregulation and sweating disorders, even in stressful situations. Patients primarily succumb to premature heart or brain damage before the age of 50. Due to the nonspecific nature of symptoms, obtaining a proper diagnosis can take several years.
Causes of Fabry Disease
Fabry Disease is classified as a rare lysosomal storage disorder. It is caused by a mutation in the GLA gene, leading to either a complete or significant reduction in the activity of the alpha-galactosidase A enzyme.
Consequently, globotriaosylceramide (GL-3) accumulates in the cells of various tissues and organs, as the enzyme deficiency prevents its breakdown. The accumulation of GL-3 disrupts the functions of affected organs and causes complications.
This rare disease is X-linked, which means it occurs more frequently in males. However, heterozygous females can also exhibit symptoms, with varying degrees of severity. In women, the clinical manifestation of the disease generally appears several years later than in men.
Diagnosis of Fabry Disease
To confirm a diagnosis of Fabry disease in men, the activity of alpha-galactosidase A is measured in plasma, leukocytes, or fibroblasts. If enzyme deficiency is detected, genetic testing is performed. In women, since alpha-galactosidase A activity can remain within normal ranges, genetic testing is necessary to identify carriers of the faulty gene responsible for Fabry disease.
Symptoms of Fabry Disease
It presents in two phenotypes:
- Classic phenotype – More common in men, characterized by absent or minimal enzyme activity, with symptoms appearing in childhood or adolescence.
- Non-classic phenotype – Symptoms appear later in life due to a lesser degree of alpha-galactosidase A deficiency.
Symptoms appearing in adolescence include:
- Intolerance to high temperatures
- Impaired sweating
- Hyperthermia
- Paresthesia (intense burning pain in the distal limbs)
- Abdominal pain, constipation, diarrhea, nausea, and vomiting
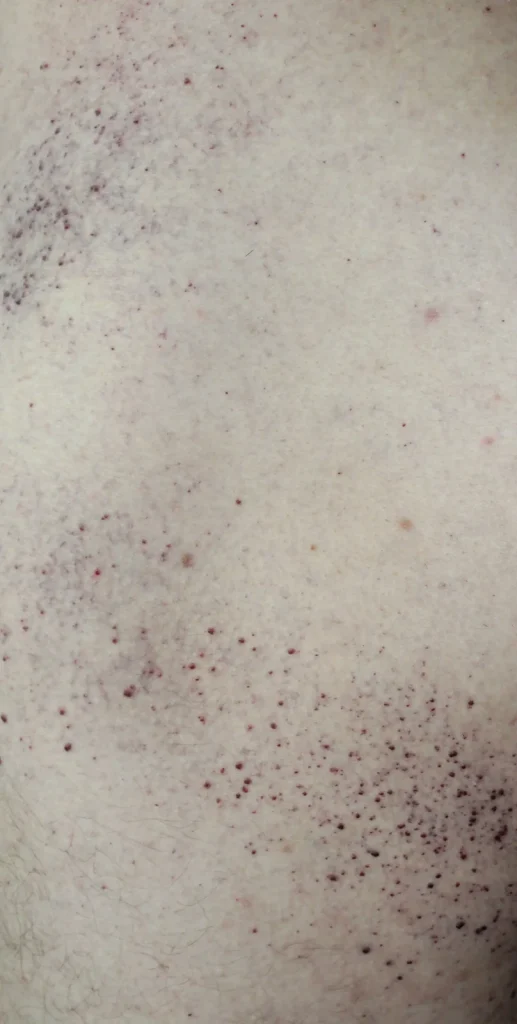
- Skin lesions on the thighs, buttocks, and lower abdomen in the form of red-purple rashes (angiokeratoma)
- Corneal clouding, cataracts, and progressive hearing loss
In adulthood, symptoms include:
- Structural and functional cardiovascular abnormalities (left ventricular hypertrophy, arrhythmias, and conduction disorders)
- Kidney damage, initially presenting as proteinuria and microalbuminuria
- Chronic fatigue, tinnitus, and dizziness
Additionally, behavioral changes such as anxiety, social isolation, depression, and feelings of guilt are common. Many of these symptoms appear before diagnosis, as Fabry patients, like those with other rare diseases, often experience long diagnostic delays, impacting both physical and mental well-being. Studies show that the quality of life in Fabry patients is significantly lower compared to the general population.
Prognosis
The most common causes of death in Fabry patients are stroke and heart attack. Compared to the general population, men with Fabry disease have a life expectancy reduced by 15-20 years, while women live approximately 6-10 years less.
Treatment Options
The standard treatment for Fabry Disease is enzyme replacement therapy (ERT) with agalsidase alfa or beta. Given the multisystemic involvement of the disease, supportive therapies are also necessary to manage complications and prevent further health deterioration.
ERT can slow disease progression and enable patients to maintain family, work, and social activities.
Additionally, migalastat has been approved for treating Fabry disease. Unlike intravenous ERT, migalastat is an oral medication. However, only patients with specific mutations responsive to this drug can receive it—approximately 35-50% of all Fabry patients.
Supportive therapies for Fabry disease include:
- Pain management, primarily through pharmacotherapy
- Neuroprotective measures (angiotensin inhibitors and angiotensin receptor blockers)
- Antiarrhythmic medications
- Pacemaker or implantable cardioverter-defibrillator (ICD) placement for severe cardiovascular involvement
- Dialysis or kidney transplantation in cases of advanced renal impairment
Treatment for Fabry disease is lifelong, and early intervention increases the chances of delaying or preventing severe complications.
Prevalence
Globally, its prevalence is estimated at 1 in 40,000 to 1 in 170,000 individuals. However, due to the complexity of Fabry disease symptoms and challenges in differential diagnosis, these figures may be underestimated.